

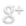


Every year over 1.2 million people suffer myocardial infarction (MI). The resulting heart damage requires new approaches for effective repair. Stem cell therapies provide hope. However none of the stem cell therapies currently in clinical trials addresses the need for efficient stem cell targeting to cardiac tissue or the need to replace efficiently dead tissue with new cardiomyocytes. To address these problems, we have built several genetic circuits that work sequentially to repair the heart. First, we have built an inducible differentiation circuit that closely resembles the endogenous differentiation pathway, to program cells to become cardiomyocytes. Second, we have built circuits that use the extracellular domains of chimeric proteins to target cells to damaged cardiac tissue. Upon binding, novel receptor-coupled intein-mediated signaling domains activate effector genes that then aid in integration, inhibition of cell death, and the alteration of the tissue microenvironment.
Introduction
There are many problems that existing therapies, and those currently in clinical trials, do not address. Some of the main issues include: how to recognize damaged heart tissue, how to deliver stem cells to areas beneath the surface, the inability of adult stem cells to differentiate into cardiomyocytes in the absence of external cues, the prevention of stem cell death during therapy, and how to stimulate pathways for new cardiac cell integration. Because all current therapies fail to address one or a combination of these problems, cell-based therapies for MI patients do not work very well and provide only modest improvement in function, if any.
In 2007 the RSI Bay Area Consortium Team designed and engineered novel methods of targeting damaged cardiomyocytes. Since then, we have shown that our targeting circuit for damaged cardiomyocytes binds and relays effectors in cultured rat cardiomyoblasts. To complement this circuit, the 2008 RSI Bay Area Consortium Team also designed and created an efficient inducible method for the differentiation of cardiomyocytes. Together, the two circuits resolve many of the problems associated with the current therapies for MI patients.
Targeting Infarcted Cardiac Tissue
To target cells efficiently to damaged cardiac tissue, the 2007 RSI Bay Area Consortium Team choose three different targets: C-reactive protein (CRP), ICAM, and myosin heavy chain. From these three targeting systems, we focused this year on continuing the characterization of the CRP based intein system.
CRP As A Target Of Infarcted Cardiac Tissue
During an MI, the inflammation causes a several fold increase in CRP serum levels as well as the generation of CRP by damaged cardiomyocytes. However, it is the deposition and immobilization of serum CRP onto exposed small nuclear ribonucleoproteins of infarcted cardiac tissue that makes it an ideal target for therapeutic applications. Because nuclear ribonucleoproteins are contained intracellularly, and only exposed upon serve cellular damage, there is minimal or no non-specific binding.
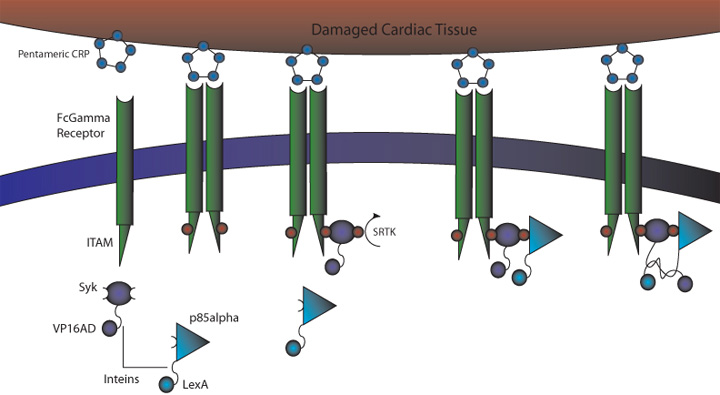
Figure 1. CRP Signaling System
1) MI induces large increase in serum CRP levels
2) CRP deposits onto damaged cardiomyocytes
3) Pre-programmed cells expressing FcGammaRIIa-R131 bind CRP on the surface of damaged cardiomyocytes and in the serum surrounding the damaged area
4) Crosslinking of the receptors induces the phosphorylation of the ITAMs in the intracellular region of the receptor
5) Syk is recruited to the receptor and becomes phosphorylated at ITAMs
6) Crosslinking of the receptors also recruits p85 which binds to the phosphorylated Tyr-317 residue on Syk
7) This interchange allows for the inteins (dnaEc-VP16AD at C terminus of Syk and dnaEn-mLexA at N terminus of p85) to interact and self-splice
8) Self splicing of the inteins produces the VP16AD-mLexA transcription factor needed for downstream effector expression

Figure 2. CRP Receptor and TF Reconstitution
FCGamma Receptor Binds Immobilised CRP on Damaged Cardiomyocytes In Vitro
As proof of principle to test targeting and how well the circuit works, we transfected human HEK293T kidney cells which transfect at very high efficiency (50-95%).
To test how well the targeting works we used the H9C2 rat cardiomyocyte cell line to model infarction. Confluent H9C2 cells were exposed to ischemic buffer for 45 minutes and incubated with 300ug/ml CRP for 2 hours. 293T cells transfected with CMV- FcGammaRIIa and a control plasmid expression DsRed2 at a molar ratio of 10:1 were removed from plates by treatment with PBS/EDTA, washed and plated on H9C2 cells. After 30 minutes cells were washed vigourously 3x and viewed with fluorescent microscopy to detect binding of FcGammaRIIa positive cells.
We observed (Fig 3 and Table 1) that FcGammaRIIa positive cells (red) bound the CRP coated H9C2 ischaemic cells at much greater affinity than FcGammaRIIa. It is important to note that cells were still capable of binding in the presence of 150ug/ml competitor CRP, which is promising for in vivo experiments given that CRP is sometimes found in the blood at nearly these levels after MI.
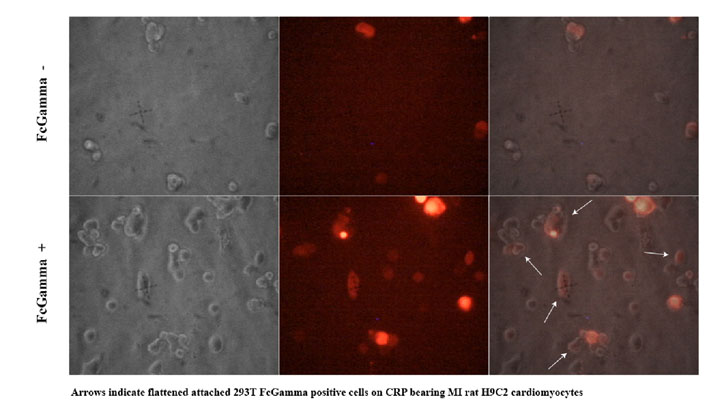
Figure 3. FcGammaRIIa containing cells bind CRP coated ischemic H9C2 cells in the presence of 150ug/ml CRP. FcGamma negative cells bind poorly.
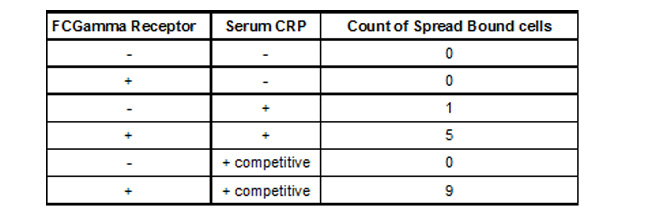 Table 1. Binding of 293T FCGamma Receptor Positive Cells To MI H9C2 with C-Reactive Protein
Table 1. Binding of 293T FCGamma Receptor Positive Cells To MI H9C2 with C-Reactive Protein
To demonstrate whether this system would work with healthy cardiomyocytes targeted to damaged cardiomyocytes, we used the ischemic system described to target normal H9C2 ectopically expressing FcGammaRII (Fig 4). FcGamma RII expressing cells bind to the ischemic H9C2 monolayer (Fig 4d), but cells carrying the control vector do not (Fig 4b). Also, H9C2 cells infected with FcGammaRII construct were able to differentiate into cardiac muscle (data not shown).
 Figure 4. H9C2 rat cardiomyocytes infected with lentiviral vector expressing dsRED2 and FcGammaRII bind monolayer of ischemic H9C2 cardiomyocytes treated with CRP. a,b) H9C2 infected with control vector. c-d) H9C2 infected with FcGammaRII. 3 red cells in d demonstrate binding to ischaemic CRP positive cardiomyocytes.
Figure 4. H9C2 rat cardiomyocytes infected with lentiviral vector expressing dsRED2 and FcGammaRII bind monolayer of ischemic H9C2 cardiomyocytes treated with CRP. a,b) H9C2 infected with control vector. c-d) H9C2 infected with FcGammaRII. 3 red cells in d demonstrate binding to ischaemic CRP positive cardiomyocytes.
CRP activates GFP signaling upon FCGamma Binding
We also tested whether CRP could activate our CRP circuit when transfected into H9C2 cells. H9C2 cells were transfected with equimolar ratios of CMV-FcGammaRIa, cmv- Syk-dnaEc-VP16, cmv- Syk-dnaEc-VP16,and LexApro-GFP reporter. DsRed2 plasmid was included at a 1:10 molar ratio to mark transfected cells. 150ug/ml CRP was added at 48 hours post-transfection. Cells were visualized 24 hours later. 4c and 4f compare the full circuit in the absence or presence of CRP. Fig. 4f shows green GFP expressing cells indicating that the circuit worked, demonstrating proof of concept.
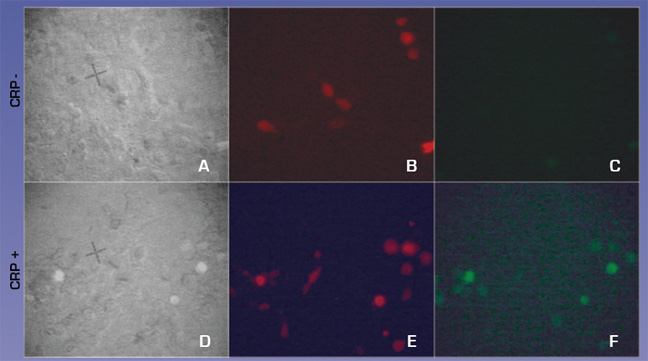
Figure 5. CRP activates the CRP circuit in HEK293T cells. A,D GFP positive cells against total cells B,E Red cells indicate CRP circuit positive cells C,F GFP positive cells indicating activation of CRP circuit
Generation of Clonal lines of H9C2-FCGammaR2a Cardiomyocytes
To characterise the affinity of the FCGammaR2a receptor we infected H9C2 cells with the FCGammaR2a lentiviral construct. 14 days post-infection we diluted H9C2-FCGammaR2a positive cells, DsRed2 positive cells, so that colonies would form from single cells. We have created 5 clonal H9C2-FCGammaR2a lines.
Fig 6. Clonal line of H9C2-FCGammaR2a cells
Effectors To Aid In Integration, Anti-apoptosis, And The Alteration Of The Tissue Microenvironment
Upon CRP binding by the receptors and the subsequent reconstitution of the split inteins, the formation of a transcription factor, LexA-VP16 is released to activate effectors to promote survival, promote angiogenesis, and discourage scarring. All downstream effectors are under the control of a lexA minimal promoter.
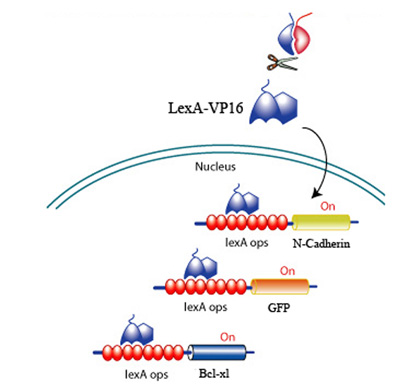
Fig 7. LexA-VP16 transcriptional activation of effectors under the control of a lexA operator
eGFP Reporter
To visualise expression of effectors, we created a enhanced green flourescent protein (eGFP) construct under the control of the lexA minimal promoter.

N-Cadherin Integration Effector
To target and replace damaged cardiomyocytes below the surface we created a effector construct with N-cadherin, a cell-cell adhesion molecule. The differential expression of N-cadherin on the surface of cells has been shown to allow cells to aggregate in different pattern formations.
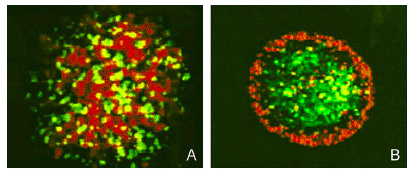
Fig 9. Sorting of Transfected L cells expressing N-Cad at 2.4 (green):1 (red) ratio. Foty and Steinberg, 2005

Fig 10. N-caherin effector construct for aid in cell integration
Anti-Apoptotic, Necrotic Effector
One of the biggest problems with current MI therapies is that most cardiomyocytes derived from stem cells die before successful integration. Therefore, we designed a set of effectors under the control of lexA-VP16 to block endogenous apoptotic and necrotic pathways. We have targeted the major apoptosis family of effector proteins, bad/bax and caspases, as well as the calpain family of proteases. In the case of non-apoptotic cell death, we added an anti-necrotic effector.
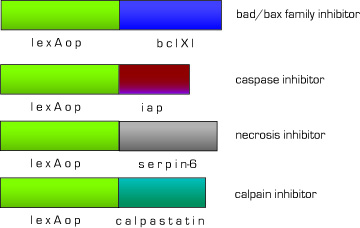
Fig 11. Anti-apoptotic,necrotic effector constructs
Angiogenic and Anti-scarring Effectors
To increase the integration of new cardiomyocytes we also created effector constructs to change the tissue microenvironment. To induce angiogenesis, the formation of new blood vessels, we created a vascular endothelial growth factor (VEGF) effector construct. This will provide new cardiomyocytes as well as nearby pre-existing non-functional hibernating cardiomyocytes with a source of oxygen and nourishment. Also, one of the major impediments to regeneration in the evolution of our immune system is the process of scarring. Although scarring plays a seminal part in the healing process, scar tissue actually inhibits and decreases cardiac ventricular function in post-MI patients. To decrease the amount of scarring and allow for cell integration before fibrotic tissue formation, we also created a hepatocyte growth factor (HGF) effector construct.
Fig 12. Angiogenesis and anti-scarring effector constructs
Advantages of the CRP Signaling System
1) Binds to CRP on damaged cells to allow for targeting of therapeutic agents (versatile since CRP is expressed in a variety of inflamed tissues)
2) Removes exposed CRP from the damaged area, lessening the damage caused by CRP and increasing the recovery of cardiac function
3) Widespread usability/adaptability for other CRP-associated pathologies such as atherosclerosis.
4) Signaling system uses endogenous pathway
5) Limited potential for cross-talk
CRP Targeting Circuit Concerns
1) FcGamma can bind serum IgG. Although the mutant version used in our system, FcGammaRIIa-R131, exhibits weak affinity for IgG, there is a possibility of binding and crosslinking of the receptors. This problem can be alleviated if the the FcGamma-CRP system is used as part of an AND gate with another receptor system, such as the ICAM-based system that we designed.
2) CRP is present at a basal level in plasma which may interfere with signaling activation of pathway. However, we have shown in preliminary experiments that cells bearing FcGammaR2a can recognize damaged cardiomyocytes even in the presence of concentrations of CRP equivalent to that found in the serum of patients with MI.
Future Directions
We have shown that cells bearing the CRP receptor system specifically bind to CRP deposited on damaged cardiomyocytes, and that the signal is then relayed to turn on expression of downstream effector genes. Our next goal is to tune the sensitivity of the FCGammaR2a receptor to minimize potential background from endogenous CRP. We intend to do this by inducing mutations in the receptor in our H9C2-FCGammaR2a clonal cell lines that we have already created. We can also attenuate the circuit sensitivity by using reduced affinity LexA mutants.
In our second generation systems we will create a more universal receptor signaling system. With the design of a single chain antibody (scFv) coupled to a “backbone” receptor signaling system similar to the one described here we could easily adapt our system to target different tissues.
Cardiomyocyte Differentiation Circuit
We designed an inducible cardiomyocyte differentiation circuit modeled after our current understanding of cardiomyocyte differentiation from studies in mouse embryonic carcinoma cell line, p19 and its derivatives, and in mouse and human stem cell lines. We use the key regulatory proteins in endogenous cardiomyocyte differentiation and have adapted it to work in parallel with our targeting circuit.
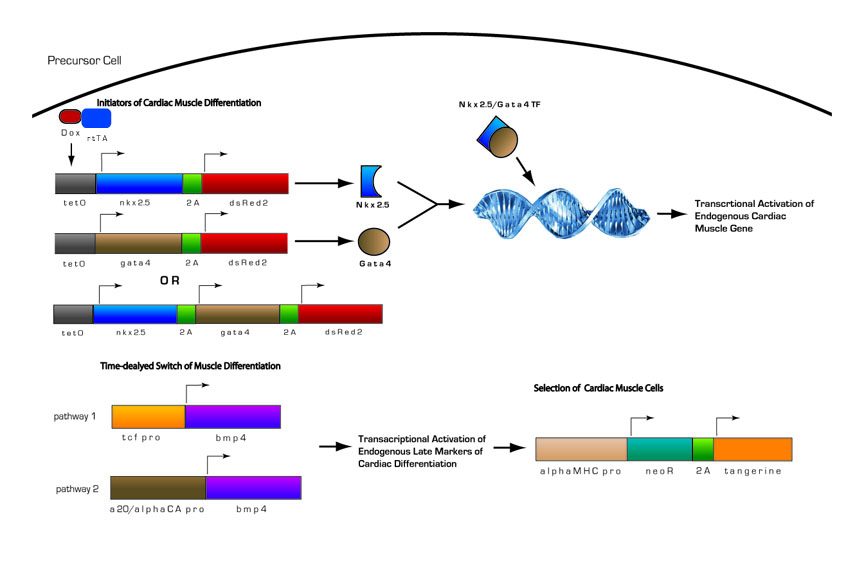
Fig 13. Inducible Cardiac Muscle Differentiation Circuit
1) Pre-programmed cells with genetic circuitry are induced to initiate cardiomyocyte differentiation by the addition of doxycycline
2) Doxycline activates transcription of two key initiators of cardiomyocyte differentiation, Nkx-2.5 and Gata4.
3) Nkx-2.5 and Gata4 act as transcriptional activators of other key regulatory proteins in cardiomyocyte differentiation. Nkx-2.5 and Gata4 activate themselves and Mef2c, another transcriptional activator, in positive feedback loops, as well as other endogenous proteins. Thus, even in the absence of doxycycline, once the circuit is induced, cells will proceed down the pathway autonomously.
4) After the cells have become determined for cardiomyocyte differentiation, Bmp4 will become activated. Bmp4 is under the control of either of two different minimal promoters, both of which provide delayed expression of Bmp4. This is needed because if Bmp4 is expressed too early in cardiomyocyte differentiation, it will have inhibit differentiation. If it expressed after commitment to the cardiomyocyte lineage it will promote differentiation
5) During late stages of cardiomyocyte differentiation, contractile proteins are expressed. A portion of the differentiated cardiomyocytes will also contain the selection construct with the neomycin resistance gene (NeoR) under the control of the myosin heavy chain promoter. With the addition of an antibiotic such as G418, we will be able to select for differentiated cardiomyocytes.
Cardiomyocyte Differentiation Pathway
Tetracycline Inducible Initiators of Cardiomyocyte Differentiation – nkx-2.5 and gata4
For real world therapeutic applications of our technology, a tightly controlled inducible system for cardiac muscle differentiation is needed. Differentiation of the cardiac progenitor cells must be regulated so that targeting and differentiation can occur in a controlled manner. Therefore, we designed both Nkx-2.5 and Gata4, early initiators of cardiomyogenesis, to be under the control of an inducible tetracycline expression system adapted from the pTRIPZ shRNAmir vector (Openbiosystems). There are two main components that comprise the pTRIPZ’s Tet-On inducible system enabling induction: the tetracycline response element (TRE) and the transactivator, rtTA. Both Nkx-2.5 and Gata4 will be under the control of the TRE promoter, a string of tetracycline operators fused to the CMV minimal promoter. The TRE exhibits reduced basal expression and tighter binding to its transactivator, rtTA, which eliminates the high background, leaky expression associated with most inducible expression systems. In the Tet-On system, expression of genes downstream of the TRE are off until exogenous doxycycline is added, thus Nkx-2.5 and Gata4 will be off until doxyxcycline is added. Nkx-2.5 and Gata4 will then activate other transcription factors necessary for cardiomyocyte differentiation. In addition, Nkx-2.5 and Gata4 will induce a positive feedback loop and activate endogenous expression of themselves so that even in the absence of doxycycline, Nkx-2.5 and Gata4 expression will continue. As a visualization marker to observe the induction of gene expression, we fused dsRed2 to the Nkx-2.5 and Gata4 constructs via the foot and mouth disease 2A sequence (fmdv2a) which allows for the expression of polycistronic messages.
Reference for 2A sequence
The Foot and Mouth Disease Virus 2A sequence for the generation of polycistronic messages from Fang, J., et al. Stable antibody expression at therapeutic levels using the peptide 2A sequence. 2005. Nat Biotech 23; 584-590. Fang et al showed that fmdv2a sequence allowed for the continuous in vivo expression of full length monoclonal antibody at therapeutic levels. 2A sequences from other viruses, such as picorna and picorna-like viruses have been used in protein cleavage of single messages.
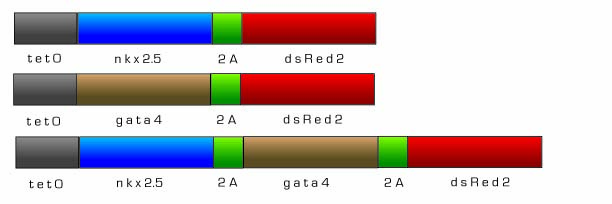
Fig 14. Tetracycline Inducible nkx-2.5 and gata4 constructs
Constitutive rtTA protein for Tet-On Inducible System
In our adapted version of the pTRIPZ tetracycline inducible, Tet-On system, we designed a constitutively active version of the transactivator, rtTA protein, under a CMV promoter, to control the expression of cardiac progenitor initiators, nkx-2.5 and gata4.
From Openbiosystems’ description of the rtTA in the Tet-On system “pTRIPZ transactivator, known as the reverse tetracycline transactivator 3 (rtTA3) binds to and activates expression from TRE promoters in the presence of doxycycline. The rtTA3 transactivator is a modified version of the wildtype in two ways. First, unlike the original tetracycline transactivator the rtTA3 is modified to bind to the TRE in the presence of doxycycline rather than in its absence. Secondly, there are three mutations within the transactivator that increase its sensitivity to doxycycline by 25 fold over the initial rtTA without increasing background activity (Das, et al. 2004). Das, et al. (2004) “Viral evolution as a tool to improve the tetracycline-regulated gene expression system” JBC Vol 279: 18, 18776-18782.”

Fig 15. CMV-rtTA construct
Delayed Expression Bmp4 Constructs
Induction of BMP4 during early stages of cardiomyocyte differentiation results in inhibitory effects. In our circuit, Nkx2.5 and Gata4 will be induced and initiate the pathways of cardiac differentiation. During normal cardiac muscle differentiation BMP4 is a downstream target of Nkx-2.5 and Gata4, the expression of which turns on at later stages of cardiac differentiation. To ensure that BMP4 has delayed expression, we designed two different BMP4 constructs under the control of minimal promoters of middle to late markers of cardiac muscle differentiation, TCF and alpha cardiac actin.
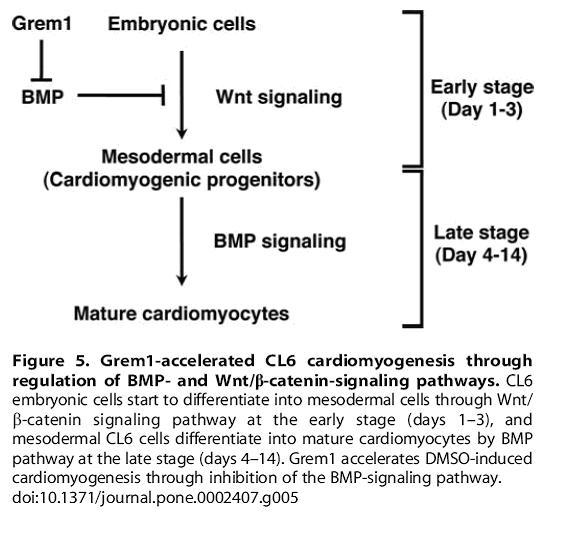
Fig 16. From Kami D, Shiojima I, Makino H, Matsumoto K, Takahashi Y, et al. (2008) Gremlin Enhances the Determined Path to Cardiomyogenesis BMP4 expression at early stages inhibits cell from entering into the cardiomyogenic pathway.
Reference for TCF minimal pro
Gaire, M., et al. Structure and expression of the human gene for the matrix metalloproteinase matrilysn.1994. JBC 269:3 2032-2040
From previous studies, we found a minimal promoter sequence where Beta-catenin/tcf4 could bind and control the expression of MMP7 “Thomas Brabletz, Andreas Jung, Serpil Dag, Falk Hlubek, and Thomas Kirchner. ?-Catenin Regulates the Expression of the Matrix Metalloproteinase-7 in Human Colorectal Cancer. 1999. American J. of Path. 155(4):1033-1038”
A20/alpha Cardiac Actin minimal promoter
Contractile proteins are expressed middle to late during cardiomyocyte differentiation making them an ideal choice as timers for the induction of BMP4. One of these cardiac specific contractile proteins is alpha cardiac actin which is one of the first contractile proteins to be expressed, making it a good choice to drive BMP4 expression. We want BMP4 expression to be delayed enough not to be inhibitory, but not so much that it fails to act in time. We have designed delayed BMP4 expression under the control of an alpha cardiac actin minimal promoter.
A20+ linker is a triple repeat of A20, a nkx2.5 consensus binding sequence that nkx2.5 binds to with high affinity. This A20 sequence was initially identified through random oligonucleotide selection as described in Chen, C.Y. and Schwartz, R.J. (1995) JBC 270(30) 15628-15633.
The alpha cardiac actin minimal promoter was described in previous work by Schwartz and collegues who revealed that the interactions of nkx2.5, serum response factor (SRF), gata4, along with other transcription factors play an important role during cardiomyocyte differentiation. One of the downstream targets of these transcription factors in cardiomyogenic pathways was alpha cardiac actin, whose promoter they identified as being under direct control of nkx2.5 as reviwed in Chen, C. and R.J. Schwartz. 1997. 11(6) 812-822.
To create a late strong minimal promoter of cardiomyogenesis we designed 3 repeats of A20 fused to the promoter sequence of cardiac alpha-actin with known cardiac specific regulation (Sepulveda, J.L., et al. GATA-4 and Nkx-2.5 Coactivate Nkx-2 DNA Binding Targets: Role for Regulating Early Cardiac Gene Expression (1998) 18(6) 3405-3415).
Fig 17. Delayed Expression Bmp4 Constructs
Alpha Cardiac Myosin Heavy Chain Minimal Promoter Screen and Selection Marker
Some contractile proteins are expressed very late in the cardiomyocyte differentiation pathway making them an ideal choice as timers for a reporter construct. One of these late-expressing cardiac specific contractile proteins is alpha cardiac myosin heavy chain. We have designed a delayed reporter, tangerine, and a selectable marker, NeoR, under the control of an alpha cardiac myosin heavy chain minimal promoter. Thus, only after the programmed cell has proceeded late into the cardiac differentiation pathway, will alpha cardiac myosin heavy chain expression be turned on and thus selection for cardiac cells can be initiated.
The neomycin resistance gene (NeoR) encodes for aminoglycoside 3‘-phosphotransferase protein. Cells that express this protein can effectively phosphorylate, and thereby inactivate, aminoglycoside antibiotics such as neomycin. This allows for the selection of mammalian cells that have a plasmid that encodes for the NeoR gene with antibiotics such as G418, geneticin (Invitrogen).

Fig 17. Alpha MHC NeoR, tangerine Construct
Differentiation Circuit Advantages
1) Our cardiomyocyte differentiation circuit does not involve exogenous pro-differentiation chemical reagents such as DMSO (other than doxycycline)
2) We utilize key endogenous transcription factors from the cardiomyogenic pathway.
3) The system is inducible.
4) Circuit timing is tightly controlled with early and delayed expression of key regulatory proteins.
5) The NeoR gene selects for pure populations of cardiomyocytes.
Differentiation Circuit Concerns
Q:Since it is known that injection of many kinds of stem cells turn cancerous in a patient, wouldn’t there be an immediate problem with using these circuits in stem cell therapy?
A: No. Differentiation of precursor cells will occur prior to targeting. Therefore after selection with an antibiotic such as G418, only cardiomyocytes will remain. And since cardiomyocytes are post-mitotic, the possibility of cancer arising from our therapy because our precursor cells are stem cell-like is very unlikely. Secondly, our system is amenable to work in many different precursor cells that need not be stem cells. From the literature, there is a good possibility that this therapy can be used on cardiofibroblasts or non-cardiac cells such as adipocytes or leukocytes via transdifferentiation.
Future Directions
We have built the differentiation circuit constructs, so our next focus is to characterize them in cultured cells. After preliminary testing in cell culture we will continue by integrating both circuits into embryonic stem cells and conduct in vivo tests in rats that have undergone an MI by coronary artery ligation. Using current techniques, we will be able to track and visualize in real time our pre-programmed cells as they target the infarcted tissues and undergo further cardiomyocyte differentiation to the adult cardiomyocyte phenotype.
Gene Therapy And Synthetic Biology
Gene therapy proponents have long sought to revolutionise the field of medicine by replacing faulty genes with healthy ones. And in 2002 their day had come, the miracle from their efforts achieved. However, their dream was short lived. It was discovered that many of the “cured” patients had developed cancer and died as a result. Thus, gene therapy has suffered a sort of dark ages, where researchers are apprehensive to try anything related to gene therapy.
Our synthetic circuits allows us to bypass the problem of cancer associated with current gene therapy techniques. Because a more intricate network can be designed into a cell, we will be able to program cells to become cardiomyocytes before human injection. The cells will be returned to the patient in a post-mitotic state and therefore have almost no chance of causing cancerous after being administered.
Use As A Novel Therapy For MI Patients
Current therapies are largely preventative: for instance, grafting-based bypass surgery, blood thinners and stents for clogged coronary arteries. Once an individual has undergone an infarction, however, current therapies are less effective. For example, some inotropic drugs can damage the kidneys and heart as they work to increase blood volume and cardiac output. The entire system may fail if the heart can not maintain homeostasis. If the damage is bad enough, the kidneys will retain more fluid volume, and overwork the heart. This will lead to more damage and, eventually, death by multi-organ failure. Furthermore, although such drugs can stimulate cardiac output, they do not fundamentally change the ultimate capability of the damaged heart to efficiently pump blood.
Those with MI often suffer permanent damage. Once the heart has undergone scarring, it can never completely heal to regain its previous level of function. Current stem cell therapy lacks sophistication and only provides a modest possibility for success. It is our hope that we may offer a therapy for these individuals, one that is not merely preventative, but can actually lead to recovery.
Parts submitted to the iGEM parts registry
Part: BBa_K080010
Construct: puc18-LexAop-eGFP-SV40PA Notes: This construct works. eGFP gives a very good signal and the lexAop has high sensitivity for the LexA transcription factor.
Team Advisor
Andrew Mendelsohn
Team Members
Jennifer Lei, UC Berkeley ’06
Joshua Resa, Stanford ’08
Sean Allen, UC Berkeley ’09
Simina Ticau, UC Berkeley ’10






 Ethereum
Ethereum Tether
Tether Xrp
Xrp Binance coin
Binance coin Litecoin
Litecoin Stellar
Stellar Bitcoin cash
Bitcoin cash Dogecoin
Dogecoin Usdcoin
Usdcoin Monero
Monero